在从基因到蛋白质的旅程中,新生的RNA分子可以在翻译成蛋白质之前以不同的方式进行切割和连接或剪接。这个过程被称为选择性剪接,允许单个基因编码几种不同的蛋白质。替代剪接发生在许多生物过程中,例如当干细胞成熟为组织特异性细胞时。然而,在疾病的背景下,替代剪接可能会失调。因此,重要的是要检查转录组 - 即可能源于基因的所有RNA分子 - 以了解疾病的根本原因。
然而,从历史上看,很难“读取”整个RNA分子,因为它们通常有数千个碱基长。相反,研究人员依赖于所谓的短读长RNA测序,它破坏RNA分子并将它们测序成更短的片段 - 大约在200到600个碱基之间,具体取决于平台和协议。然后使用计算机程序重建RNA分子的完整序列。
短读长RNA测序可以提供高度准确的测序数据,每个碱基的错误率低至约0.1%(这意味着每测序1,000个碱基错误地确定一个碱基)。然而,由于测序读取的长度较短,它可以提供的信息有限。在许多方面,短读长RNA测序就像将一张大图片分成许多形状和大小相同的拼图,然后试图将图片重新拼凑在一起。
最近,可以端到端测序长度超过10,000个碱基的RNA分子的“长读长”平台已经可用。这些平台不需要在测序之前分解RNA分子,但它们具有更高的每个碱基错误率,通常在5%到20%之间。这一众所周知的限制严重阻碍了长读长RNA测序的广泛采用。特别是,高错误率使得难以确定在特定条件或疾病中发现的新的,以前未知的RNA分子的有效性。
为了解决这个问题,费城儿童医院(CHOP)的研究人员开发了一种新的计算工具,可以从这些容易出错的长读长RNA测序数据中更准确地发现和量化RNA分子。该工具名为ESPRESSO(错误统计PRomoted Evaluator of Splice Site Options),今天在Science Advances上报道。
“长读长RNA测序是一项强大的技术,将使我们能够发现罕见遗传疾病和其他疾病(如癌症)中的RNA变异,”CHOP计算和基因组医学中心主任Yi Xing博士说。
“我们可能正处于如何发现和分析RNA分子的拐点。从短读长RNA测序到长读长RNA测序的转变代表着令人兴奋的技术变革,迫切需要能够可靠地解释长读长RNA测序数据的计算工具。
ESPRESSO可以仅使用容易出错的长读长RNA测序数据,就可以准确发现和定量来自同一基因(称为RNA亚型)的不同RNA分子。为此,计算工具将给定基因的所有长RNA测序读数与其相应的基因组DNA进行比较,然后使用单个长读段的错误模式来自信地识别剪接连接 - 新生RNA分子被切割和连接的地方 - 以及它们相应的全长RNA亚型。
通过查找长RNA测序读数和基因组DNA之间的完美匹配区域,以及借用基因的所有长RNA测序读数的信息,该工具能够识别高度可靠的剪接连接和RNA亚型,包括以前未在现有数据库中记录的那些。
研究人员使用模拟数据和真实生物样本数据评估了ESPRESSO的性能。他们发现ESPRESSO在发现RNA亚型和量化它们方面都比目前可用的多种工具表现更好。研究人员还生成并分析了超过10亿个长RNA测序读数,涵盖30种人体组织类型和三种人类细胞系,为研究全长RNA亚型分辨率下的人类转录组变异提供了有用的资源。
“ESPRESSO解决了长期存在的长读长RNA测序问题,并可能迎来新的发现机会,”Xing博士说。“我们设想ESPRESSO将成为研究人员在各种生物医学和临床环境中探索细胞RNA库的有用工具。
智能推荐
-

开发用于智能基础设施系统的自感知超材料混凝土2023-08-23 混凝土是建筑行业中最常用的材料,其历史可以追溯到罗马帝国。匹兹堡大学的工程师们现在正在重新构想21世纪的设计
-
新的药物递送方法利用凝血靶向肿瘤2023-08-25 威斯康星大学麦迪逊分校的研究人员开发了一种通过利用血小板的凝血倾向来靶向肿瘤的新方法。29月<>日,《科学进展》杂志首次描述了这种新方法,它增加了威斯
-
研究人员提出了新的对流理论 用于理解电池的快速充电2023-08-22 MechSE副教授KyleSmith和博士生MdAbdulHamid最近在《电源杂志》上发表了一篇文章
-
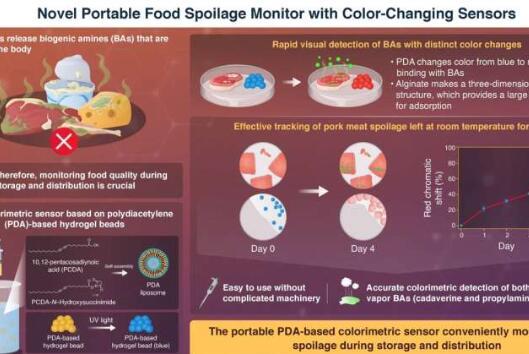
研究人员开发便携式变色食品腐败变质传感器2023-08-24 当鱼、肉和奶酪等食物分解时,它们会释放出各种低分子量有机氮化合物,称为生物胺(BA)。虽然身体在激素合成等过程中少量使用BA,但从变质的食物中摄入大量BA会导致严
-

巫师之旅还没结束!曝《霍格沃茨之遗》续作开发中2023-09-13 据insider-gaming讯息,《霍格沃茨之遗2》正在开发中,宛如仍由AvalancheSoftware开发
-

研究人员开发出不受气体和液体影响的弹性材料2023-08-05 一个国际研究小组开发了一种技术,该技术使用液态金属来制造一种不受气体和液体影响的弹性材料。该材料的应用包括用作需要气体防护的高价值技术的包装,例如柔性电池
-

《虫子快餐店》开发商正在进入全新作品开发阶段2023-08-19 曾开发过冒险游戏作品《虫子快餐店》的工作室YoungHorses此刻已经开始为其全新项目进行预热。YoungHorses合伙创始人兼总裁PhilipTibitos
-
研究人员发现新的超强酸2023-08-01 帕德博恩大学的研究人员成功地生产了非常特殊的催化剂,称为“刘易斯超强酸”,可用于破坏强化学键并加速反应
-

校本课程开发的核心(校本课程开发的意义)2023-08-06 大家好,小范来为大家解答以上的问题。校本课程开发的核心,校本课程开发的意义这个很多人还不知道,现在让我们一起来看看吧
-

研究人员对奇怪金属的神秘领域有了新的见解2023-08-27 所谓的“奇怪金属”的行为长期以来一直困扰着科学家们,但多伦多大学的一组研究人员可能离理解这些材料更近了一步
-
研究人员开发有史以来第一个通过激素调节来控制食欲的可摄入的电子装置2023-08-26 纽约大学阿布扎比分校(NYUAD)的一组研究人员,由纽约大学Tandon生物工程助理教授兼纽约大学阿布扎比分校高级神经工程和转化医学实验室主任KhalilRama
-

12000年前的岩刻让研究人员感到困惑 暗示文明已经失落2023-08-10 位于南美洲秘鲁的一个偏远地区发现了一批神秘的岩刻,这些岩刻的年代可以追溯到公元前12年左右。这批岩刻的惊人之处在于它们描绘了一些现代人类认为不可能在那个时期存在的